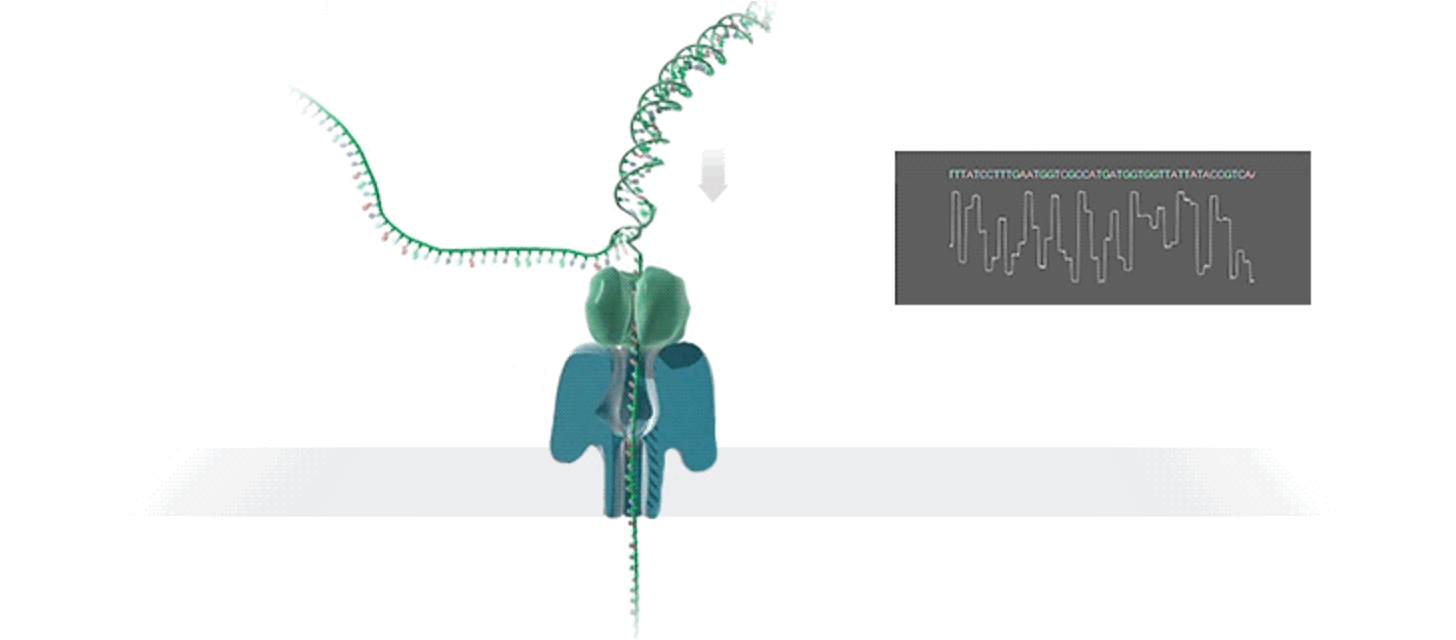
Nanopore sequencing: leveraging an emerging technology
The development of culture-independent analysis of microbiomes was enabled by so-called next-generation sequencing (Bennett, 2004, Margulies et al., 2005, Bentley et al., 2008, McKernan et al., 2009). Illumina was the most broadly used sequencing platform of 2010s, and revolutionized the life sciences by reducing the cost of sequencing by orders of magnitude, providing exceptionally accurate sequencing data, and increasing throughput. However, the latest generation of emerging sequencing technologies (i.e., “third generation sequencing”) is in in the process of disrupting biomedical research once more (Athanasopoulou et al., 2021). One particular technology that has gained considerable traction in the last several years is nanopore sequencing (Oxford Nanopore Technologies, Inc. [ONT]) (Jain et al., 2015). While Illumina sequencing-by-synthesis technology generates sequences that are generally 150 or 300 base pairs of DNA in length, ONT sequencing theoretically has no upper limit on the length of output sequence, with single reads of over 1 million base pairs of DNA routinely reported (Jain et al., 2015). Because most genomes contain repeat regions spanning much longer than 300 base pairs, the puzzle that is a given genome cannot typically be completed with Illumina short-read sequencing alone (i.e., because all these short reads match up nonspecifically to all their cognate sequences in the repeat regions of the chromosome). The long reads produced by ONT sequencing much more easily span the repeat regions of genomes enabling significantly easier assembly of complete chromosomes. However, until recently ONT technology was plagued with a considerably higher error rate than competing technologies, limiting its applicability (Amarasinghe et al., 2020). This error rate decreased in recent years, with a crucial inflection point being reached in 2022, when studies illustrated that the contemporary ONT instrumentation and software could produce genome assemblies with an error rate on par with Illumina sequencing (Sereika et al., 2022).
The Baker Lab was an early adapter of ONT technology, and in 2022 published the first protocols to obtain multiple complete bacterial genomes simultaneously directly from saliva using ONT sequencing (Baker, 2022). Several of the genomes completed using these methods were the first complete genome for their given species (Candidatus Saccharibacteria* HMT-870, Candidatus Saccharibacteria* HMT-348, Actinomyces graevenitzii) (Baker, 2021, Baker, 2022, Baker, 2022). Particularly intriguing were the genomes from the enigmatic Saccharibacteria family, which are essentially tiny (even by bacterial standards) parasitic bacteria that depend on larger host bacteria to survive (He et al., 2015). The novel complete genomes from our study illustrated that the G6 group of Saccharibacteria likely has a different lifestyle, and possibly host and host dependencies than the more well-understood G1 group of Saccharibacteria (Baker, 2021). These differences likely extend to ecological and pathogenic roles as well, as Saccharibacteria appear to have a relationship to inflammation and periodontal disease (albeit poorly understood at this stage) (Chipashvili et al., 2021). The ability to obtain complete genomes directly from complex samples, such as saliva, will revolutionize microbiology research, as it was previously only possible to obtain complete genomes of species that were isolated and cultivated in the lab in a pure culture (i.e., only incomplete, draft genomes could be obtained from metagenomes using earlier sequencing technologies)(Athanasopoulou et al., 2021). Obtaining genomes that are both complete and accurate is of importance because they then allow accurate identification and quantification of the species of interest in microbiome samples, and enable accurate prediction of the metabolic capabilities, and therefore ecological and pathogenic roles of the species (Venter et al., 2004, Naito et al., 2016). This data can further guide wet-lab research and help scientists design experiments, isolate, cultivate, and study species that were previously intractable (Cross et al., 2019). In addition to genomics and metagenomics, The Baker Lab pioneered use of the ONT sequencing platform for RNA sequencing of oral bacteria (Baker et al., 2022). RNA sequencing via ONT has several advantages as well. Because ONT can sequence native DNA and RNA molecules (unlike most sequencing methods, which must first reverse transcribe the RNA to cDNA, and/or amplify the DNA or RNA with PCR), ONT sequencing can detect base modifications, such as methylation, as well as noncanonical bases such as inosine (Garalde et al., 2018, Tourancheau et al., 2021, Begik et al., 2022, Nguyen et al., 2022). The ability to detect DNA and RNA modifications on a genome wide or transcriptome wide scale is a major advance and is likely produce entire new fields of microbiology research. Furthermore, the long reads enable the detection of co-transcribed genes and RNA isoforms on a transcriptome wide scale (Garalde et al., 2018, Grunberger et al., 2022). As none of the OHSU core facilities currently provide nanopore sequencing, The Baker Lab is eager to collaborate or consult on OHSU projects that could benefit from an ONT approach.